

La Protoporfiria Eritropoietica è una patologia comunemente definita “allergia al sole”, appartenente ad una famiglia di disordini metabolici causati da difetti nell’attività di uno degli enzimi della via biosintetica dell’eme, in questo specifico caso, parliamo di una mutazione autosomica recessiva del gene che codifica per la ferrochelatasi (FECH), ovvero l’enzima terminale di questa “via”.
Ad oggi, la Protoporfiria Eritropoietica (o EPP) risulta essere la terza patologia più diffusa all’interno di questa famiglia, pertanto: conosciamola meglio.
QUALI SONO LE CAUSE DI EPP?
L’EPP rientra nel campo delle cosiddette “malattie rare” e perlopiù è di origine ereditaria. Ciò che la caratterizza a livello biologico è un accumulo di Protoporfirine fototossiche dato appunto dall’assenza di FECH, a livello di plasma sanguigno, eritrociti e tessuti, che si vanno a depositare in particolar modo in pelle e fegato, manifestando a livello cutaneo una fotosensibilità dolorosa acuta, eritema, edema, prurito e sensazioni di bruciore in seguito all’esposizione solare.
Possiamo parlare di episodi di gravità variabile a seconda della durata dell’esposizione al sole, che possono arrivare a provocare lesioni croniche permanenti sulla superficie cutanea esposta, soprattutto a livello delle zone più sensibili.
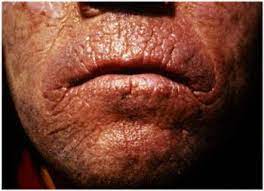
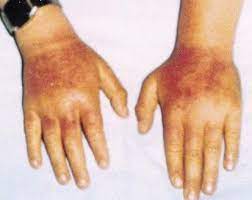
Nei casi più gravi, come accennavamo in precedenza, possono venire escreti attraverso la bile, causando calcoli biliari, fibrosi, con episodi ostruttivi e malattie epatiche croniche che potrebbero evolvere in insufficienza epatica acuta in breve tempo.
Non è da escludere, a questo proposito, la necessità di un trapianto di fegato, qualora si verifichino gravi complicanze.
COME RICONOSCERLA?
L’EPP solitamente si manifesta con i primi sintomi già in età neonatale per durare poi, tutta la vita.
Sono comuni ustioni cutanee in seguito anche ad una breve esposizione al sole, tuttavia, la gravità della patologia può essere differente a seconda del soggetto, del tempo di esposizione e di conseguenza possono variare anche le forme fisiche con cui si palesa.
Potrebbero, infatti, non apparire immediatamente manifestazioni cutanee visibili, ma unicamente una sensazione dolorifica acuta percepita dal paziente. In questo caso, ad esempio, il neonato sarebbe portato a piangere ininterrottamente, rendendo difficile la comprensione della patologia stessa da parte degli adulti.
Il dolore in questione può risultare così insopportabile da causare anche nei pazienti adulti episodi di forte nervosismo, tensione ed aggressività.
Manifestazioni cutanee, invece, se trascurate possono dare origine a croste e cronicizzare, ispessendo la cute.
DIAGNOSI: DALLA SUA SCOPERTA, AD OGGI.
L’EPP è stata scoperta per la prima volta nel 1961.
Inizialmente la sua diagnosi risultava complicata – basti pensare che negli ultimi 20 anni era stata mancata in 9 pazienti su 10 – tuttavia, ad oggi ci sono stati notevoli progressi nella sua comprensione e prognosi grazie all’utilizzo, tra le altre cose, di una strumentazione innovativa, come il microscopio a fluorescenza.
Nello specifico, questa patologia viene stabilita rilevando un aumento dei livelli di Protoporfirina nel plasma e nei globuli rossi e rilevando un picco di fluorescenza plasmatica a 634 nm.
La protoporfirina eritrocitaria deve, inoltre, essere frazionata per determinare le proporzioni di Protoporfirina contenenti o meno zinco: nell’EPP la percentuale di Porfirina priva di metalli è quasi sempre superiore all’85%.
Ulteriori test che possono essere utilizzati per la sua diagnosi, sono i test genetici per le mutazioni di FECH o ALAS 2.
Pazienti che presentano parenti con questo tipo di anomalia genica risultano essere più a rischio di manifestare la patologia.
È di comune riscontro, seppur non sintomo di EPP, inoltre, una carenza di Vitamina D, mentre sporadicamente possono verificarsi anche casi di anemia lieve da carenza di ferro.
COME GESTIRLA?
La gestione di questa patologia include primariamente la prevenzione, da effettuare proteggendo la pelle dall’esposizione solare.
Come? Riducendo, ad esempio, l’attività all’aperto o utilizzando indumenti protettivi e creme a base di biossido di titanio o ossido di zinco, in grado di assorbire i raggi UV e fungere dunque da protezione nei confronti dei raggi solari.
Tuttavia, come si può evincere facilmente, la necessità di riadattare la propria vita potrebbe influire negativamente sulle attività sociali e lavorative della persona e, in generale, sulla qualità della vita stessa.
Ad oggi, la somministrazione di beta-carotene per via orale è un’ulteriore strategia utilizzata per migliorare, almeno lievemente, la tolleranza al sole, grazie sia al buon quantitativo di antiossidanti utili a neutralizzare i radicali tossici, sia alla possibilità di dare una colorazione “preventiva” alla pelle, aumentandone la pigmentazione e riducendone la penetrazione della luce responsabile dello sviluppo di sintomi cutanei.
Affine al beta-carotene è la somministrazione di Alfamelanotide, ovvero un analogo dell’ormone umano in grado di stimolare gli alfa-melanociti ed aumentare il pigmento della melanina, producendo una costante “abbronzatura” nel paziente e riducendo la fotoattivazione. Oltre a ciò, sembra inoltre in grado di migliorare i processi di riparazione del DNA, modulando appunto l’infiammazione.
Sono stati condotti, a questo proposito, studi clinici che hanno mostrato un un miglioramento della qualità della vita riferito dai pazienti.
Come abbiamo precedentemente detto, inoltre, nei casi più gravi un organo bersaglio dell’EPP risulta essere il fegato, responsabile dell’eliminazione della Porfirina attraverso il sistema epatobiliare. In casi di degenerazione di questo sistema, potrebbe essere richiesto un trapianto di organo.
LA SCIENZA è IN CONTINUO SVILUPPO
La “fame” da parte della scienza di trovare una soluzione definitiva all’EPP non finisce quì.
Sono in corso, infatti, ulteriori studi ed esperimenti che sembrano offrire risultati rincuoranti.
Nello specifico: come abbiamo detto all’inizio, l’eccessivo accumulo di Protoporfirina risulta essere la problematica centrale. A questo fattore è correlata la produzione di radicali liberi ed i conseguenti eventuali danni a livello della pelle.
Ma come viene trasportata nel circolo ematico l’EPP? Il suo efflusso sembra essere mediato dal trasportatore ABCG2, aiutato appunto dall’esposizione ai raggi UV.
La conoscenza di questi dati, pertanto, ha portato gli scienziati ad ipotizzare che la soppressione di questo trasportatore (ABCG2), potrebbe portare a ridurre la fototossicità e l’epatotossicità in modo permanente.
Al fine di validare questa ipotesi, sono stati recentemente condotti esprimenti sui topi da cui sono emersi risultati del tutto rincuoranti: gli animali a cui è stata soppressa l’attività di ABCG2 hanno riscontrato un significativo aumento di PPIX nei globuli rossi, ma ridotto nel plasma e nella pelle, con conseguente attenuazione di fototossicità cutanea.
Anche a livello di epatotossicità ulteriori analisi hanno confermato la possibilità di ridurre i livelli di PPIX nel sistema biliare, prevenendo il suo blocco, sempre grazie alla riduzione di ABCG2.
Sono in corso ulteriori studi per validare definitivamente questa possibilità, per eliminare l’idea che il trasportatore possa essere sostituito da un analogo, ed appurare una concreta possibilità di eseguire questo trattamento con successo anche nell’uomo.
Fortunatamente sembrano esserci buone possibilità.
Referenze
– Todd DJ, Nesbitt GS, Lavery TD, Trimble ER, Burrows D. Erythropoietic protoporphyria. The problem of a suitable screening test. Acta Derm Venereol. 1990;70(4):347-50. PMID: 1977265.
– Linenberger M, Fertrin KY. Updates on the diagnosis and management of the most common hereditary porphyrias: AIP and EPP. Hematology Am Soc Hematol Educ Program. 2020 Dec 4;2020(1):400-410. doi: 10.1182/hematology.2020000124. PMID: 33275677; PMCID: PMC7727547.
– Sarkany RP. Erythropoietic protoporphyria (EPP) at 40. Where are we now? Photodermatol Photoimmunol Photomed. 2002 Jun;18(3):147-52. doi: 10.1034/j.1600-0781.2002.00708.x. PMID: 12207680.
– Wang P, Sachar M, Lu J, Shehu AI, Zhu J, Chen J, Liu K, Anderson KE, Xie W, Gonzalez FJ, Klaassen CD, Ma X. The essential role of the transporter ABCG2 in the pathophysiology of erythropoietic protoporphyria. Sci Adv. 2019 Sep 18;5(9):eaaw6127. doi: 10.1126/sciadv.aaw6127. PMID: 31555729; PMCID: PMC6750912.
– Erwin AL, Balwani M. Porphyrias in the Age of Targeted Therapies. Diagnostics (Basel). 2021 Sep 29;11(10):1795. doi: 10.3390/diagnostics11101795. PMID: 34679493; PMCID: PMC8534485.
AUTORE: Greta Oldani Biologa e Freelance di Scienze Salute e Benessere.

